R.用于传播可重复研究的包
2019年12月26日
内容
最后修改:2019年5月31日
1摘要
本教程适用于所有想要写作的人R.包。R.是开发用于分析和理解现实世界数据的新统计方法的一种极好的语言。R.包提供了一种在可重复的、文档化的单元中捕获新方法的方法。一个R.包非常容易创建,创建一个R.打包有很多好处。在本教程中,我们创建一个R.包中。我们从一个数据集和一个简单的脚本开始,以一种有用的方式转换数据;也许您有自己的数据集和脚本?我们将脚本替换为函数,将功能和数据放入一个R.包。然后添加文档,以便我们的用户(以及将来的自己)理解函数的功能以及如何将函数应用于新数据集。与一个R.包裹在手中,我们可以解决更多的提高挑战:小插曲对于包装的丰富叙事描述;单元测试让我们的包装更加强大;和版本控制记录我们如何更改包。开发我们的包的最后一步是与他人共享它,通过github,通过CRAN,或者通过特定领域的通道,如生物体。
2生物学和统计学动机
2.1测定单细胞上的基因表达:单细胞RNA序列
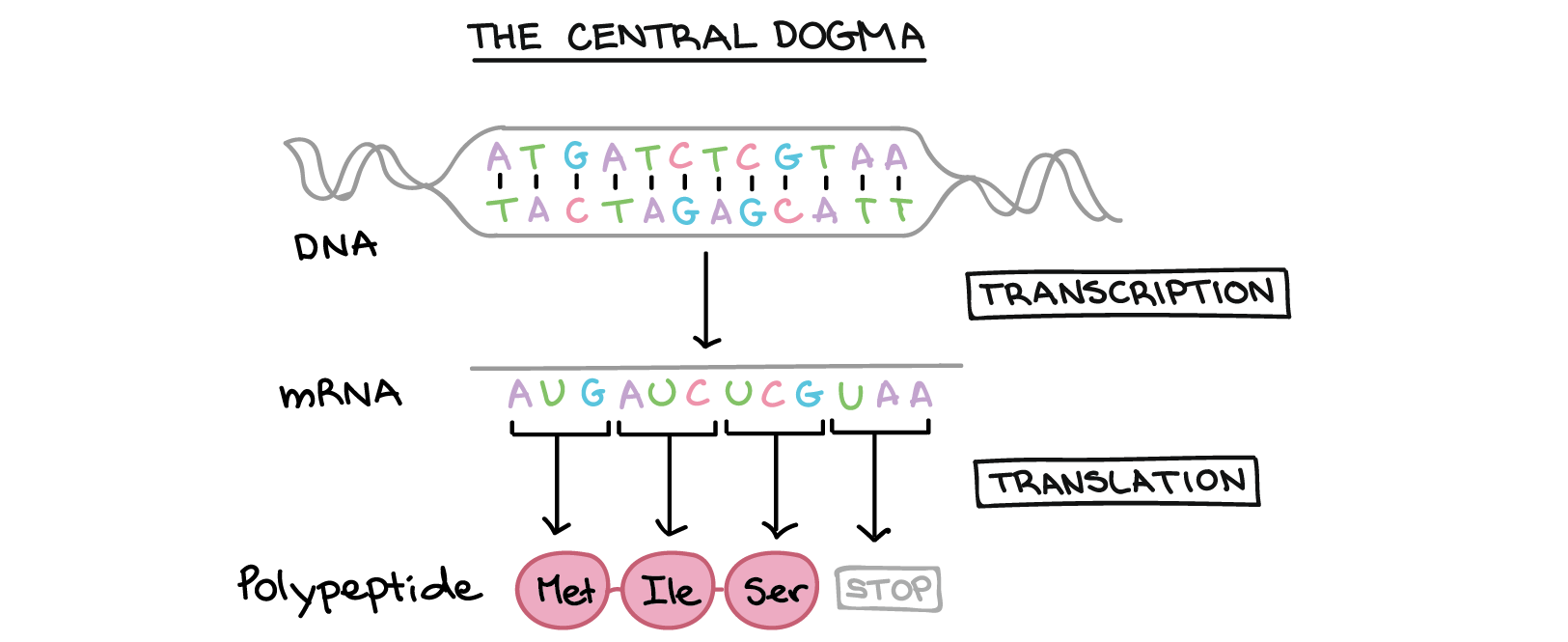
分子生物学的“中央教条”:在DNA(染色体)中编码的基因被转录为mRNA,然后翻译成突出菌。

- https://cdn.kastatic.org/ka-perseus-images/2b597889d05bc601803a3b4d9ec5ccd5e7b8d3af.png可汗学院的所有内容都可以在www.khanacademy.org免费获得
{kind=link}
RNA测序(散装RNA-SEQ)
- 从大型细胞样品中分离mRNA
- 逆转录到cDNA,片段和序列
- 对齐测序片段参照基因组
- 更多的片段对齐被解释为更高的基因表达。

单细胞RNA-seq
- 分离单个细胞
- 将每个细胞与条形码珠子联系起来
- 条形码cDNA序列
- 大多数目前的方法导致非常稀疏的“覆盖范围”

- Hwang等人,2018https://doi.org/10.1038/s12276-018-0071-8
2.2模拟数据
参数:
N_GENES < - 20000 N_CELLS < - 100 ##伽马分布式基因意味着速率< - .1形< - .1 ##负二氯计数分散< - 0.1非常粗略的模拟:
set.seed (123) gene_means < - rgamma (n_genes形状=形状,率=)cell_size_factors < - 2 ^ rnorm (n_cells, sd = 0.5) cell_means < -外(gene_means cell_size_factors,‘*’)计数< -矩阵(rnbinom (n_genes * n_cells,μ= cell_means大小= 1 /分散),nrow = n_genes ncol = n_cells)模拟数据的基本性质
范围(计数)## [1] 0 256 ##零的均值(counts == 0)## [1] 0.793133 ##'库大小' - 读取每个单元座位(COLSUMS(计数),main =“图书馆大小”)
##每种基因沟槽的平均排污(Rowmeans(log1p(counts)),main =“log基因表达”)
2.3大小的因素
log_counts < - log(counts)居中< - log_counts - rowmeans(log_counts)filtered_median < - function(x)中位数(x [是finite(x)])size_factors < - exp(应用(居中,2,filtered_median))(size_factors)
## [1] 0.4438464 2.8639456 median(size_factors) ## [1] 1.0257813.最基本的
3.1R.包
磁盘上的文件和目录集合。完整包可能具有如下所示的结构。
SCSimulate DESCRIPTION NAMESPACE R/模拟。R size_factors。R.男子/simulate.Rd size_factors.Rd vignettes/ Using_this_package.Rmd tests/ testthat.R testthat/ test_simulate.R test_size_factor.R描述
- 包名称,标题,描述(如纸摘要)
- 作者和维护者(联系作者)
- 许可证
这个包依赖的其他包('依赖项')
取决于:使用此包所需的数据结构或工作流程。进口:在当前包装中使用。例如,我们将使用功能rgamma ()那rnorm(),rnbinom ()从统计包中。建议:用于举例或小插图。
名称空间
- 这个包使用的函数-
进口()那importFrom () - 该封装的函数可以为用户提供 -
出口()
r /
- 文本文件包含R.函数定义
男子/
- 文本文件记录功能
小插曲/
- “Markdown”或其他描述软件包使用的文本文件。
测试/
- 用于为包提供“单位”的功能。
3.2包骨架
创建一个包
devtools::创建(SCSimulate) # #✔创建‘SCSimulate / # #✔活动项目设置为' /用户/ ma38727 / b / github / BiocIntro /小插曲SCSimulate”创建的R / # # # #✔✔写作“描述”# #包:SCSimulate # #标题:什么包(一行,标题)# #版本:0.0.0.9000 # # Authors@R(解析):## * First Last [aut, cre] (YOUR-ORCID-ID) ##描述:包做什么(一段)。## code: UTF-8 ## LazyData: true ## ## write 'NAMESPACE' ## Setting active project to '' 编辑描述文件(纯文本)
标题:模拟单细胞RNA seq数据版本:0.0.0.9000 Authors@R: c(person(given = "Martin", family = "Morgan", role = c("aut", "cre"), email = "Martin.Morgan@RoswellPark.org", comment = c(ORCID = " you -ORCID- id ")), person(given = "Another", family = "Author", role = "aut"))该包使用基因表达值的γ分布和每个细胞计数的负二项模型模拟单细胞RNA序列数据。该包还包含预处理函数,包括库缩放因子的简单计算。导入:stats编码:UTF-8 LazyData: true到目前为止,我们的包裹看起来
SCSimulate DESCRIPTION NAMESPACE R/ .名称空间3.3从脚本到函数
将描述模拟的脚本的一部分转换为函数模拟()。使用函数参数来捕获默认值。
模拟<函数(n_genes = 20000,n_cells = 100,速率= 0.1,形状= 0.1,色散= 0.1){gene_means < - rgamma(n_genes,shape = shape,rate = rate)cell_size_factors < - 2 ^ rnorm(n_cells,sd = 0.5)Cell_means < - 外部(Gene_means,Cell_size_factors,`*`)矩阵(rnbinom(n_genes * n_cells,mu = cell_means,size = 1 /色散),nrow = n_genes,ncol = n_cells)}将描述大小因子计算的脚本部分转换为函数size_factors ()。唯一的论点size_factors ()是一个计数矩阵。
.filtered_median <- function(x [is.finite(x)]) size_factors <- function(counts) {log_counts <- log(counts) centered <- log_counts - rowMeans(log_counts) exp(apply(centered, 2, .filtered_median))}}检查一下我们没出什么差错。
set.seed(123)计数< - simulate()size_factors < - size_factors(counts)范围(size_factors)## [1] 0.8639456中位数(size_factors)## [1] 1.0257813.4向包中添加函数定义
控件中的文件中放置函数r /目录中。通常,以文件中的函数/函数组命名文件。例如,
文件:R /模拟。R.
模拟<函数(n_genes = 20000,n_cells = 100,速率= 0.1,形状= 0.1,色散= 0.1){gene_means < - rgamma(n_genes,shape = shape,rate = rate)cell_size_factors < - 2 ^ rnorm(n_cells,sd = 0.5)Cell_means < - 外部(Gene_means,Cell_size_factors,`*`)矩阵(rnbinom(n_genes * n_cells,mu = cell_means,size = 1 /色散),nrow = n_genes,ncol = n_cells)}文件:R / size_factors。R.
.filtered_median <- function(x [is.finite(x)]) size_factors <- function(counts) {log_counts <- log(counts) centered <- log_counts - rowMeans(log_counts) exp(apply(centered, 2, .filtered_median))}}我们的包裹现在看起来像
SCSimulate DESCRIPTION NAMESPACE R/模拟。R size_factors。R.3.5文档的功能
使用Roxygen2.为文档添加标记行#'在每个函数的上面。常见标记如下所示。
@title是出现在帮助页面顶部的一行描述。@描述在标题之后提供函数的简短描述。使用@details更多的描述出现在“用法”部分(生成基于签名后的功能@export帮助页面)。- 文件参数(
@param)及归还(@return仔细)值。的@param值用于形成帮助页的“Arguments”部分。的@return值将出现在帮助页的“返回”部分中。 @例子都包含在帮助页面的“Examples”部分,并且必须是完整的、语法正确的R代码(在构建和检查包时计算示例)。- 使用
@importFrom表示特定包提供当前包中使用的特定功能。 - 为了可读性,“换行”到80列。一致且大方地使用缩进和空格。
文件:R /模拟。R.
#' @title模拟单细胞数据#' #' @description ' Simulate() '生成基因x细胞计数矩阵#'模拟单细胞RNA-seq数据。基因表达使用gamma分布进行建模。计数使用#'负二项分布进行模拟。#' #' @param n_genes integer(1)要模拟的基因(行)的数量。#' #' @param n_cells integer(1)要模拟的单元格(列)的数量。@param rate数值(1)rgamma()分布的速率参数。@param shape numeric(1) ' rgamma() '分布的形状参数。@param dispersion numeric(1) size (' 1 / dispersion ') parameter #' the ' rnbinom() '分布。#' #' @return ' simulate()返回' n_genes x n_cells ' matrix of #'模拟单细胞RNA-seq计数。#“#”@examples # '计数< -模拟()#“暗(计数)#”的意思是(数量= = 0)# ' 0 '细胞的一部分#的范围(计数)# #“@importFrom统计rgamma rnorm rnbinom # ' # ' @export模拟< -函数(n_genes = 20000, n_cells = 100 = 0.1, = 0.1,色散= 0.1){gene_means < rgamma (n_genes形状=形状, rate = rate) cell_size_factors <- 2 ^ rnorm(n_cells, sd = 0.5) cell_means <- outer(gene_means, cell_size_factors, `*`) matrix( rnbinom(n_genes * n_cells, mu = cell_means, size = 1 / dispersion), nrow = n_genes, ncol = n_cells ) }文件:R / size_factors。R.
# ` @importFrom stats median .filtered_median <- function(x) median(x[is.finite(x)]) # ` @title计算几何均值为中心的中位数缩放细胞缩放# `因子。#' #' @description ' size_factors() '按日志计数的行平均值将每一行的日志计数居中。然后使用有限居中的#'值来计算列级几何中值#'缩放因子。#' #' @param计数matrix()基因x细胞RNA-seq计数。#' #' @return ' size_factors() '返回缩放因子的' numeric(ncol(counts)) ' vector #'。#' #' @example #' set.seed(123) #' counts <- simulate() #' size_factors <- size_factors(counts) #大约1 #' #' @export size_factors <- function(counts) {log_counts <- log(counts) centered <- log_counts - rowMeans(log_counts) exp(apply(centered, 2, .filtered_median))}}3.6更新' NAMESPACE '和' man '页
DevTools :: Document(“Scsimulate”)##更新SCSIMULED文档##编写命名空间##加载SCSIMULATE ##编写命名空间更新NAMESPACE文件
- 此包使用的函数(
统计数据::rgamma ()那统计数据::rnorm ()那统计数据::rnbinom ())。 - 指示由此包定义的旨在对用户可见的函数(
模拟()和size_factors (),但不.filtered_median ())。
- 此包使用的函数(
将上面介绍的文档转换为独立文件
- 例如,
人/模拟。理查德·道金斯 - 一个纯文本文件,但带有可被理解的latex风格标记R.。
- 例如,
我们的包裹现在看起来像
SCSimulate DESCRIPTION NAMESPACE R/模拟。R size_factors。R.男子/simulate.Rd size_factors.RdNAMESPACE文件更新为
cat(readLines("SCSimulate/NAMESPACE"), sep="\n") ## #由roxygen2生成:不手动编辑## ## export(simulate) ## export(size_factors) ## importFrom(stats,median) ## importFrom(stats,rnbinom) ## importFrom(stats,rnorm) ## importFrom(stats,rnbinom) ## importFrom(stats,rnorm)3.7建立和检查
- 构建(整理)包文件到一个“tar”球,
SCSimulate_0.0.0.9000.tar.gz。 - 检查焦油球是否完整和正确。
##写NAMESPACE ##加载SCSimulate ##写NAMESPACE ##写size_factors。理查德·道金斯## ── Building ────────────────────────────────────────────────────── SCSimulate ── ## Setting env vars: ## ● CFLAGS : -Wall -pedantic -fdiagnostics-color=always ## ● CXXFLAGS : -Wall -pedantic -fdiagnostics-color=always ## ● CXX11FLAGS: -Wall -pedantic -fdiagnostics-color=always ## ──────────────────────────────────────────────────────────────────────────────── ## ✔ checking for file ‘/Users/ma38727/a/github/BiocIntro/vignettes/SCSimulate/DESCRIPTION’ ## ─ preparing ‘SCSimulate’: ## ✔ checking DESCRIPTION meta-information ## ─ checking for LF line-endings in source and make files and shell scripts ## ─ checking for empty or unneeded directories ## ─ building ‘SCSimulate_0.0.0.9000.tar.gz’ ## ## ── Checking ────────────────────────────────────────────────────── SCSimulate ── ## Setting env vars: ## ● _R_CHECK_CRAN_INCOMING_USE_ASPELL_: TRUE ## ● _R_CHECK_CRAN_INCOMING_REMOTE_ : FALSE ## ● _R_CHECK_CRAN_INCOMING_ : FALSE ## ● _R_CHECK_FORCE_SUGGESTS_ : FALSE ## ── R CMD check ───────────────────────────────────────────────────────────────── ## Bioconductor version 3.11 (BiocManager 1.30.10), ?BiocManager::install for help ## ─ using log directory '/private/var/folders/yn/gmsh_22s2c55v816r6d51fx1tnyl61/T/Rtmp6S4exQ/SCSimulate.Rcheck' ## ─ using R Under development (unstable) (2019-12-01 r77489) ## ─ using platform: x86_64-apple-darwin17.7.0 (64-bit) ## ─ using session charset: UTF-8 ## ─ using options '--no-manual --as-cran' ## ✔ checking for file 'SCSimulate/DESCRIPTION' ## ─ this is package 'SCSimulate' version '0.0.0.9000' ## ─ package encoding: UTF-8 ## ✔ checking package namespace information ## ✔ checking package dependencies (3.4s) ## ✔ checking if this is a source package ## ✔ checking if there is a namespace ## ✔ checking for executable files ## ✔ checking for hidden files and directories ## ✔ checking for portable file names ## ✔ checking for sufficient/correct file permissions ## ✔ checking serialization versions ## ✔ checking whether package 'SCSimulate' can be installed (1.8s) ## ✔ checking installed package size ## ✔ checking package directory ## ✔ checking for future file timestamps (505ms) ## ✔ checking DESCRIPTION meta-information ## ✔ checking top-level files ## ✔ checking for left-over files ## ✔ checking index information ## ✔ checking package subdirectories ## ✔ checking R files for non-ASCII characters ## ✔ checking R files for syntax errors ## ✔ checking whether the package can be loaded ## ✔ checking whether the package can be loaded with stated dependencies ## ✔ checking whether the package can be unloaded cleanly ## ✔ checking whether the namespace can be loaded with stated dependencies ## ✔ checking whether the namespace can be unloaded cleanly ## ✔ checking loading without being on the library search path ## ✔ checking dependencies in R code ## ✔ checking S3 generic/method consistency (651ms) ## ✔ checking replacement functions ## ✔ checking foreign function calls ## ✔ checking R code for possible problems (2.1s) ## ✔ checking Rd files ## ✔ checking Rd metadata ## ✔ checking Rd line widths ## ✔ checking Rd cross-references ## ✔ checking for missing documentation entries ## ✔ checking for code/documentation mismatches (407ms) ## ✔ checking Rd \usage sections (792ms) ## ✔ checking Rd contents ## ✔ checking for unstated dependencies in examples ## ✔ checking examples (1.7s) ## ✔ checking for non-standard things in the check directory ## ✔ checking for detritus in the temp directory ## ## ## ── R CMD check results ────────────────────────────── SCSimulate 0.0.0.9000 ──── ## Duration: 14.9s ## ## 0 errors ✔ | 0 warnings ✔ | 0 notes ✔3.8安装
devtools::install("SCSimulate") ## ✔ checking for file ‘/Users/ma38727/a/github/BiocIntro/vignettes/SCSimulate/DESCRIPTION’ ## ─ preparing ‘SCSimulate’: ## ✔ checking DESCRIPTION meta-information ## ─ checking for LF line-endings in source and make files and shell scripts ## ─ checking for empty or unneeded directories ## ─ building ‘SCSimulate_0.0.0.9000.tar.gz’ ## ## Running /Users/ma38727/bin/R-devel/bin/R CMD INSTALL \ ## /var/folders/yn/gmsh_22s2c55v816r6d51fx1tnyl61/T//Rtmp6S4exQ/SCSimulate_0.0.0.9000.tar.gz \ ## --install-tests ## * installing to library ‘/Users/ma38727/Library/R/4.0/Bioc/3.11/library’ ## * installing *source* package ‘SCSimulate’ ... ## ** using staged installation ## ** R ## ** byte-compile and prepare package for lazy loading ## ** help ## *** installing help indices ## ** building package indices ## ** testing if installed package can be loaded from temporary location ## ** testing if installed package can be loaded from final location ## ** testing if installed package keeps a record of temporary installation path ## * DONE (SCSimulate)3.9使用!
图书馆(Scsimulate)模拟? size_factors示例(size_factors) ## ## sz_fct> set.seed(123) ## ## sz_fct> counts <- simulate() ## ## sz_fct> size_factors <- size_factors(counts) ## ## sz_fct> median(size_factors) # approximately 1 ## [1] 1.0257814.先进的挑战
4.1数据集
4.2沟通:雕刻叶
4.3稳健:测试和示例
4.4成熟:版本控制
4.5传播
5.sessioninfo()
sessionInfo() ## R Under development(不稳定)(2019-12-01 r77489) ## Platform: x86_64-apple-darwin17.7.0 (64-bit) ## Running Under: macOS High Sierra 10.13.6 ## ## Matrix products: default ## BLAS: /Users/ma38727/bin/R-devel/lib/libRblas。/ user /ma38727/bin/R-devel/lib/libRlapack。dylib # # # #语言环境:# # [1]en_US.UTF-8 / en_US.UTF-8 en_US.UTF-8 / C / en_US.UTF-8 / en_US。其他附加包:## [1]SCSimulate_0.0.0.9000 biocstyle_1 .15.2 ## ##通过命名空间加载(和没有附加):# # # # [1] Rcpp_1.0.3 compiler_4.0.0 BiocManager_1.30.10 [4] prettyunits_1.0.2 remotes_2.1.0 tools_4.0.0 # # [7] testthat_2.3.1 digest_0.6.23 pkgbuild_1.0.6 # # [10] pkgload_1.0.2 evaluate_0.14 memoise_1.1.0 # # [13] rlang_0.4.2 rstudioapi_0.10 cli_2.0.0 # # [16] yaml_2.2.0 xopen_1.0.0 xfun_0.11 # # [19] xml2_1.2.2 roxygen2_7.0.2 withr_2.1.2 # # [22]stringr_1.4.0 knitr_1.26 desc_1.2.0 # # [25] fs_1.3.1 devtools_2.2.1 rprojroot_1.3-2 # # [28] glue_1.3.1 R6_2.4.1 processx_3.4.1 # # [31] fansi_0.4.0 rcmdcheck_1.3.3 rmarkdown_2.0 # # [34] bookdown_0.16 sessioninfo_1.1.1 purrr_0.3.3 # # [37] whisker_0.4 callr_3.4.0 magrittr_1.5 # # [40] codetools_0.2-16 clisymbols_1.2.0 backports_1.1.5 # # [43] ps_1.3.0Ellipsis_0.3.0 htmltools_0.4.0 ## [46] usethis_1.5.1 assertthat_0.2.1 stringi_1.4.3 ## [49] crayon_1.3.4